Checking Mutation Status in scRNA-seq Data¶
Sometimes, it's useful to look for known mutations in your scRNA-seq reads, especially if you expect a mutation to be present in certain populations and not others, e.g. an inducible model system or a recurrent tumor versus a diagnostic one.
This process is relatively simple, though somewhat annoying. Let us delve into that annoyance now.
A Note of Warning
Note that this process is subject to high amounts of ambiguity. Given the sparsity of most scRNA-seq datasets, there may not be any reads for a given variant, especially if the gene is not highly expressed. Even if there is coverage, a WT call does not mean that the record is actually WT - it may only have a single read, so the other allele could well be mutant. Point being, you can really only tell if there is any support for the alternate allele in a given cell, not that the cell is WT.
The Mutation(s)¶
In this case, I only care about one mutation, so I handmade my VCF for the H3.3K27M mutation for mouse (mm10):
Ultimately, you just need a VCF of mutations derived from the same reference as your single cell data. If you have matched bulk WGS or WES data, variants called from that can be used. You can call variants from the scRNA-seq data itself, but it isn't recommended and won't be covered here. Use the GATK RNA-seq variant discovery best practices for that.
This file needs to be bgzipped and indexed via tabix as well.
The Single Cell Dataset¶
For your scRNA-seq dataset, you need aligned reads in BAM format, position sorted. Acquire these however you want, STARsolo, CellRanger, whatever. CellRanger BAMs will work by default.
You also need a list of cell barcodes in the dataset - the barcodes.tsv.gz file output by CellRanger works.
Using Vartrix¶
vartrix is a tool built by 10X to genotype specific sites in scRNA-seq data for known mutations.
Install¶
Via conda:
Running¶
Pretty simple to run. Ya just feed in your VCF, the bam, the genome, the barcodes, and an output name. Below is a script to submit LSF jobs for a directory containing CellRanger runs. I use the raw feature barcodes, as I am doing my own cell calling with alevin-fry counts for my downstream analyses, so I don't want any of my potential cells filtered out.
#!/bin/bash
module load conda3/202105
conda activate vartrix
for f in ./cellranger_v6/*; do
echo "$f";
samp=$(basename "$f")
bsub -P dnb -J vartrix_$samp -q standard -n 4 -R "rusage[mem=4000] span[hosts=1]" vartrix \
-v k27m.vcf.gz \
-b "$f"/outs/possorted_genome_bam.bam \
-f ./refdata-gex-mm10-2020-A/fasta/genome.fa \
-c "$f"/outs/raw_feature_bc_matrix/barcodes.tsv.gz \
-o ./vartrix/"$samp".K27M_vartrix.mtx
done
Edit as needed, change to full paths, whatever. Run the above script via bash vartrix.sh assuming you saved it as such. It should be run from the directory above the one containing all your CellRanger runs.
Using the Output¶
This will spit out .mtx files for each sample, which we likely want to get into R and slapped onto our single cell object. I use Bioconductor packages, so my object is a SingleCellExperiment. Seurat will be similar, and the vartrix Github has an example of how to pull stuff out. I know I only have one variant, so I just want to tack it onto my SCE's colData.
# Get Sample files.
samp.files <- list.files("./10X_scRNA_v3/processed/vartrix",
recursive = FALSE)
samp.ids <- c("P14K-1","P14K-2","P14K-3","P14K-4",
"P14-1","P14-2","P14-3","P14-4",
"P21K-1","P21K-2","P21K-3","P21K-4",
"P21-1", "P21-2","P21-3","P21-4",
"P4K-4","P4K-2","P4K-3","P4K-1",
"P4-2","P4-3","P4-1")
first <- TRUE
all.k27m <- data.frame()
for (samp in seq_along(samp.files)) {
mm <- readMM(paste0("./10X_scRNA_v3/processed/vartrix/", samp.files[samp]))
sbase <- unlist(strsplit(samp.files[samp], ".", fixed = TRUE))[1]
bars <- read.table(paste0("./10X_scRNA_v3/processed/cellranger_v6/", sbase,
"/outs/raw_feature_bc_matrix/barcodes.tsv.gz"),
header = FALSE)
mm <- as.data.frame(mm)
colnames(mm) <- bars$V1
colnames(mm) <- paste0(samp.ids[samp], "_", colnames(mm))
if (first) {
all.k27m <- mm
first <- FALSE
} else {
all.k27m <- cbind(all.k27m, mm)
}
}
Check that all our cells in our SCE object named sce are found.
Which they are. So now add the calls to the metadata.
rownames(all.k27m) <- "chr1_180811844"
all.k27m <- all.k27m[,colnames(sce)]
all.k27m <- as.data.frame(t(all.k27m))
# No reads detected
all.k27m$V1 <- stringr::str_replace(as.character(all.k27m$V1), "0", "Unknown")
# Only ref detected
all.k27m$V1 <- stringr::str_replace(as.character(all.k27m$V1), "1", "Ref_Only")
# Only alt detected
all.k27m$V1 <- stringr::str_replace(as.character(all.k27m$V1), "2", "Mut_Only")
# Both alleles detected
all.k27m$V1 <- stringr::str_replace(as.character(all.k27m$V1), "3", "Mut_WT")
sce$H3K27M_gt <- all.k27m$V1
Great, now my SCE contains a column containing the genotype info, so we can plot our calls.
dittoDimPlot(sce, "H3K27M_gt", reduction.use = paste0("UMAP_m.dist0.2_n.neigh10"),
do.raster = TRUE, color.panel = c("red", "orange", "black", "grey80"),
order = "decreasing", size = 0.5) + theme(aspect.ratio = 1)
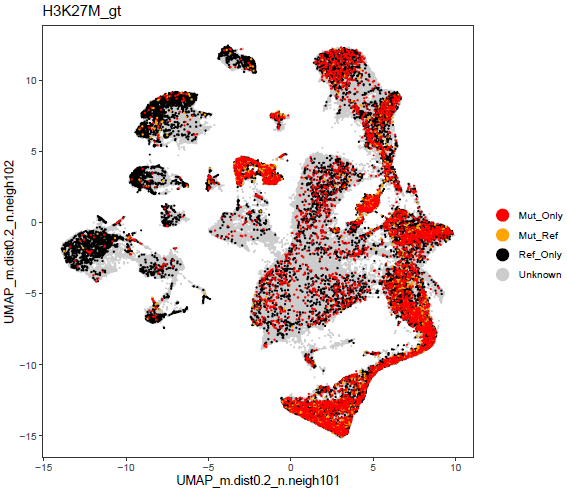
Looks pretty decent. See a few pops that didn't get much induction, and some that did, which was to be expected.